Qu’est-ce que la bêta-thalassémie ?
La bêta-thalassémie est une maladie due à une anomalie génétique de l’hémoglobine. Celle-ci est constituée de quatre « briques », aussi appelées chaines, assemblées entre elles : deux briques de type alpha et deux briques de type bêta. En cas de bêta-thalassémie, ce sont les chaines bêta qui, produites en quantité insuffisante, entrainent une production insuffisante d’hémoglobine. Certaines formes de bêta-thalassémie n’engendrent aucun symptôme tandis que des formes plus sévères entrainent des anémies, parfois extrêmement importantes.
Comment se transmet la bêta-thalassémie ?
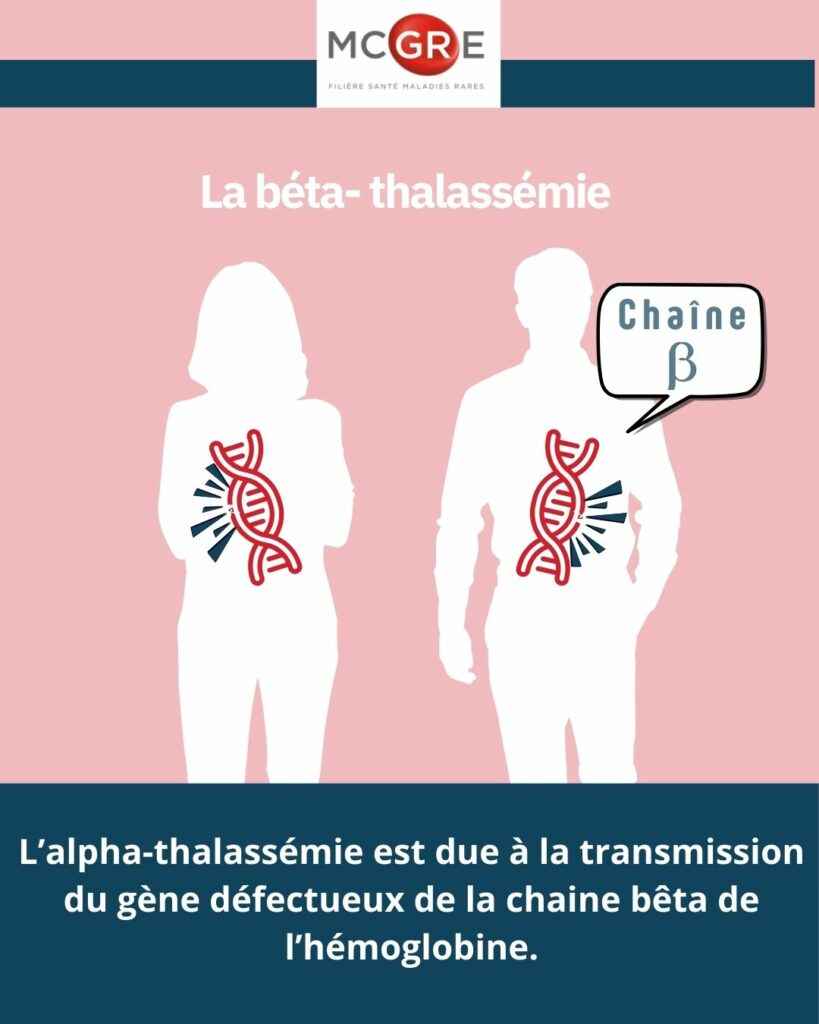
La bêta-thalassémie est une maladie héréditaire transmise à la fois par le père et par la mère. Un enfant est malade si chacun de ses deux parents lui transmet son gène défectueux de la chaine bêta de l’hémoglobine.
Quels sont les symptômes de la bêta-thalassémie ?
Les symptômes dépendent de la sévérité de la maladie :
Bêta-thalassémie mineure : cette forme de la maladie n’a généralement pas de conséquence sur la santé car le taux d’hémoglobine est normal ou proche de la normale grâce à l’autre gène, capable de produire des chaines bêta en quantité suffisante.
Bêta-thalassémie intermédiaire : pour cette forme de la maladie, bien que les deux gènes bêta soient atteints de l’anomalie, l’hémoglobine est tout de même produite en quantité réduite. Ainsi, les patients sont atteints d’anémie mais celle-ci est légère donc bien tolérée par les malades.
Bêta-thalassémie majeure : les deux gènes bêta sont altérés, rendant la production d’hémoglobine insuffisante voire nulle. Le symptôme principal est l’apparition d’une anémie progressive, entraînant une sensation de faiblesse à l’effort et une grande fatigabilité. La croissance générale s’arrête et des déformations osseuses très stigmatisantes s’installent également. Par ailleurs, en cas d’anémie aggravée, le foie et la rate augmentent de volume et provoquent un « très gros ventre ». En cas d’absence de traitement, la survie est rapidement compromise. Les patients atteints de bêta-thalassémie majeure sont également plus sensibles aux risques d’infections.
Quelle est la prise en charge de la bêta-thalassémie ?
La prise en charge repose principalement sur la transfusion sanguine afin de corriger l’anémie, combinée à un traitement chélateur pour traiter la surcharge en fer.
La splénectomie, opération consistant à retirer la rate, est conseillée lorsque le volume de celle-ci augmente.
Une prise quotidienne d’acide folique (vitamine B9), intervenant dans la fabrication des globules rouges, est également recommandée.
La greffe de moelle osseuse est le traitement curatif de la bêta-thalassémie majeure.
En cours d’étude, la thérapie génique a permis de traiter un petit nombre de patients, qui ont alors pu se passer de transfusions.

Comment établir un diagnostic de bêta-thalassémie ?
Le dépistage peut se faire dès le début de la grossesse ou à la naissance afin de prévenir les éventuelles complications. Un diagnostic peut être établi à partir d’une simple prise de sang, prescrite par le médecin, lorsqu’un enfant présente des symptômes d’anémie.
Une carte de soins et d’urgences, personnelle, peut vous être délivrée par un médecin. Elle contient des informations essentielles pour la prise en charge en urgence.